What is Spinal Muscular Atrophy?
Spinal Muscular Atrophy (SMA) refers to a group of rare, genetically inherited neuromuscular diseases that damage and kill specialized nerve cells in the spinal cord called the lower motor neurons (anterior horn cells) leading to progressive muscle weakness and atrophy. Motor neurons control movement in the arms, legs, face, chest, throat, and tongue, as well as skeletal muscle activity including speaking, walking, swallowing, and breathing.
The word atrophy implies wasting away or decreasing in size. With SMA, certain muscles become smaller and weaker due to lack of use.
The incidence of SMA is often cited as being approximately 10 in 100,000 live births. A recent review found estimates ranging from 5.0 to 24 in 100,000 births. SMA affects males and females equally. It mostly affects infants and children but can also affect adults.
Types of SMA
SMA is divided into four clinical subtypes that vary in age of onset, highest motor milestone achieved, and prognosis. While the subtypes provide a convenient means of classifying patients, it should be noted that patients exist along a continuum of disease severity with an overlap in symptoms between subtypes.
In general, earlier disease onset is associated with more severe symptoms and a lower probability of achieving motor milestones.
Current clinical classification of SMA types:
| SMA Type | Usual age of symptoms onset | Impact of muscle weakness on sitting / walking |
|---|---|---|
| Type 1 | Younger than 6 months | Unable to sit or roll independently |
| Type 2 | 6-18 months | Able to sit, but not walk, independently |
| Type 3a | 18 months-3 years | Able to walk, though may lose this ability over time |
| Type 3b | 3 years-18 years | Able to walk, though may lose this ability over time |
| Type 4 | Over 18 years | Mild walking difficulties |
Adapted from Finkel et al., 2017
Clinical presentation
SMA symptoms vary depending on the type. In general, people with SMA experience a progressive loss of muscle control, movement, and strength. Muscle loss worsens with time. The disease tends to severely affect the muscles closest to the torso and neck, leading to proximal muscle weakness. Some people with SMA never get to walk, sit, or stand while others gradually lose their ability to do these actions.
SMA type 1, also known as infantile SMA or Werdnig-Hoffmann disease, is the most common type of SMA. It affects approximately 60% of infants born with SMA and is the most severe form of the disease. Infants with SMA type 1 usually appear normal at birth but experience severe weakness before 6 months of age. Developmentally they do not achieve independent sitting and may achieve very few developmental motor milestones. Because of lower motor neuron loss, affected infants have poor suck and swallow reflexes and respiratory muscle weakness. Without intervention, affected children die before 2 years of age due to progressive respiratory muscle weakness and respiratory failure.
SMA type 2, also known as intermediate SMA or Dubowitz disease, affects about 30% of infants born with SMA. Symptoms usually appear between 6 and 18 months of age. Affected children can sit independently at some point in their development. However, this ability is usually lost by the mid-teens or later and affected individuals never achieve independent standing and walking. Additional associated symptoms include difficulty swallowing (dysphagia) and respiratory difficulties. Trembling (tremor) of the fingers is also common. In addition, weakness of the muscles supporting the spine leads to curvature of the spine (scoliosis). The life expectancy is reduced in patients with SMA type 2 but many reach adulthood.
SMA type 3, also known as juvenile SMA or Kugelberg-Welander disease, accounts for about 10% of infants born with SMA. The age of onset is variable and can be as early as 18 months or as late as the teenage years. Although affected individuals have hip and leg weakness and may fall frequently, they are able to walk independently at some point in their development. However, the ability to walk and stand may be lost as they grow and with disease progression, and many become wheelchair dependent. Long-term prognosis depends on the degree of motor function attained as a child, and respiratory muscle weakness is typically mild or absent. SMA type 3 is associated with a normal life expectancy.
SMA type 4, also known as late-onset SMA, occurs in less than 1% of people with SMA. Symptoms are less severe than in other subtypes and onset typically occurs in adulthood and most commonly after 35 years of age. All motor developmental milestones are achieved and most individuals with SMA type 4 can walk throughout their life. Patients with SMA type 4 have a normal life expectancy.
Diagnosis
Symptoms that usually lead to an SMA diagnosis are problems or delays related to muscle wasting (atrophy), which leads to difficulties with activities such as rolling over, sitting up, crawling, standing, walking, breathing, swallowing, and head and neck control.
Early diagnosis allows early monitoring and treatment, which seem to improve outcomes for people with SMA. Some research suggests that early treatment—when treatment begins before symptoms develop—improves motor outcomes and lowers the risk of death or needing a ventilator in people with SMA.
SMA can be diagnosed by prenatal testing, genetic testing, or newborn screening. If a doctor suspects SMA, a simple blood test can identify 95% of all cases. A patient may be referred to a neurologist to help detect or rule out the remaining 5% of rarer genetic mutations.
Management: A multidisciplinary approach
A multidisciplinary team is critical for children with SMA to ensure optimal outcomes keeping in mind all aspects of treatment. SMA is a severe genetic condition that requires precise diagnosis and extensive physiotherapy treatment to protect the muscles from rapid deterioration and development of contractures.
The management of SMA must be entrusted to a broad multi-disciplinary team that should provide rehabilitation, spinal management, orthopedics, and nutritional and gastrointestinal management services.
The multidisciplinary treatment approach of SMA:
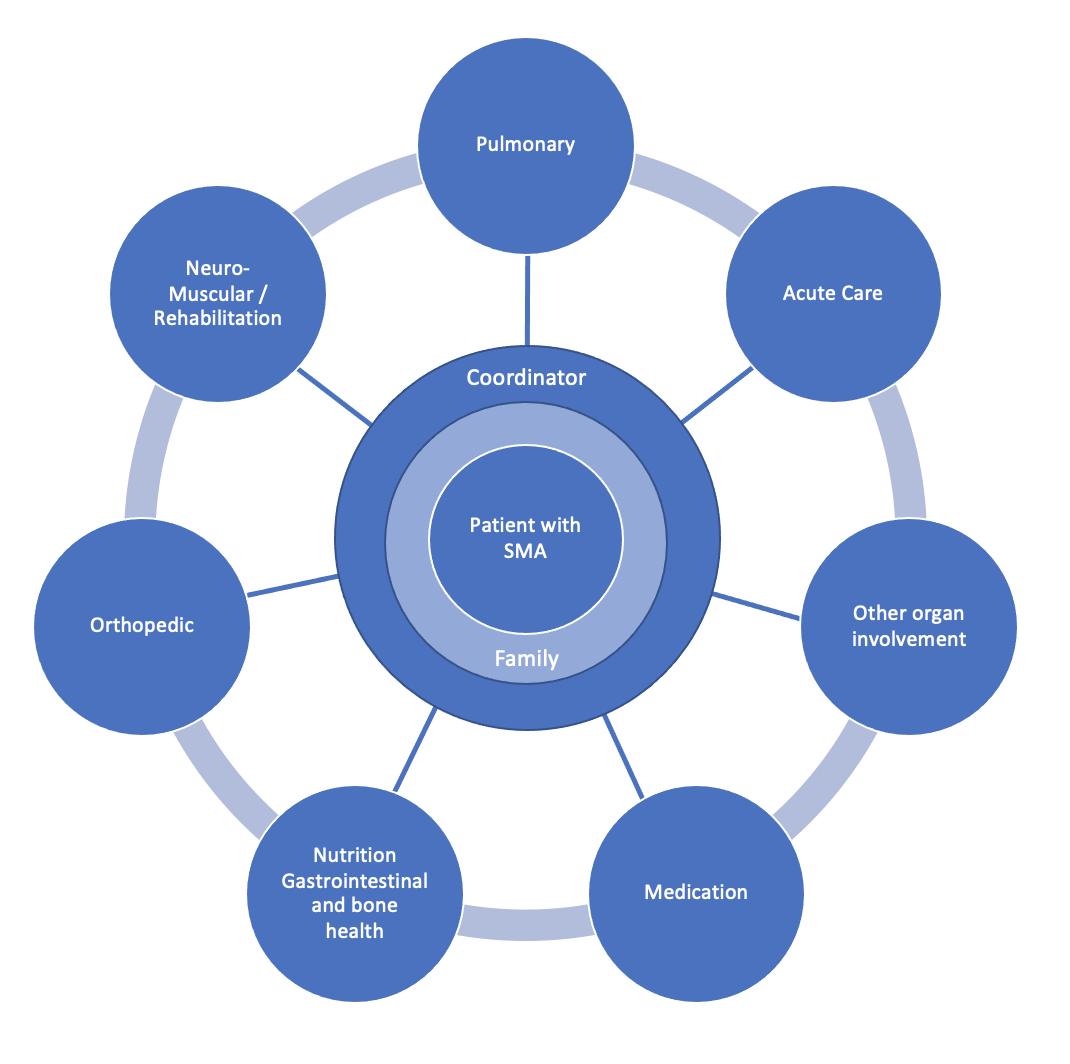
From Mercuri Eugino et al, 2018
Medical management
The most effective medical treatment for SMA 1 patients is single-dose gene replacement therapy, which improves motor functions, enhances cardiovascular functions, prevents the need for ventilation, and increases life expectancy (Mendell J 2017, Samiah A 2019).
Multiple techniques can also be followed to help manage SMA.
-
Postural management
- Sleep: Positioning systems are recommended to maintain body symmetry, prevent scoliosis, enhance sleep, and prevent discomfort and pressure on joints and to maintain short muscles in a lengthened position. For examples, frog leg position can be prevented by using braces called night splints, rolls, and cushions.
- Seating: Individuals benefit from a customized seating system such as manual or powered wheelchairs, electric scooters, and tricycles to promote postural control, respiratory function, and optimize hand function and access to leisure activities to maximize participation in daily life.
- Standing: Standers are commonly prescribed to increase weight-bearing and upright positioning to positively influence bone mineral density, range of motion, hip stability and bowel function. Prone stander or supine standers are recommended depending on the individual abilities of the child to turn head, tolerance to supported standing, and trunk control. Recommendations are made to provide more opportunities for participation in activities at home and at school.
-
Orthotics
A timely provision of orthosis such as Ankle-Foot Orthotics and Knee-Ankle-Foot Orthotics, following assessment by the physical therapists and orthopedic doctor, can provide support and alignment in ambulant and non-ambulant individuals.
Spinal braces are usually prescribed by doctors for scoliosis. The brace could aid in improving stability of spine and trunk in sitting and to improve upper limb function. Night splints are also recommended by occupational therapists to maintain upper limb positioning.
-
Mobility
Participation is a primary goal for therapists working in pediatric rehabilitation. The use of wheelchairs and power mobility has been demonstrated to enhance participation, engagement in meaningful life experiences, and social relationships for children with mobility limitations.
Manual wheelchairs, powered wheelchairs, gait trainers, and walking aids are typically recommended based on the functional levels of the child’s ability to sit, stand, walk, and use their upper limbs.
-
Assistive technology
The use of augmentative and alternative communication (AAC) for individuals with severe and progressive speech conditions can promote expressive language and means of communication with peers, teachers, family, and caregivers.
-
Environmental factors
Social, attitudinal, physical, and other environmental factors can have a significant impact on children’s mobility.
- Home environmental changes include bathroom adaptations such as grab bars, wall ladders, adjustable toilet seat and sink heights, benches in showers, space under cabinets for wheelchairs, adjustable bed heights, and widened doorways.
- School environmental adaptations include extra aid for writing, recreational-based physical education classes, access to elevators, accessible seating in classroom, tailored extracurricular activities, and group games to help meet the social interests of the child.
-
Pulmonary management
A respiratory physical therapist should be involved in assessment and management of pulmonary complications. This may include airway clearance, suctioning, postural drainage and breathing exercises to maximize oxygen capacity of the lungs.
Conclusion
SMA management approaches should consider the whole ecosystem surrounding the child. By keeping children active and engaged, goals across all the International Classification of Functioning, Disability and Health domains can be achieved (World Health Organization, 2007).
Activities and interventions for children with SMA should support the entire family, be culturally sensitive, and appropriate to the age of the child and their interests. All interventions must be child-directed, and, for children who are mostly at home, caregiver-delivered. They need to focus on including the child in the family’s existing routines or promoting new routines in which they would like to engage. Fitness goals can often be addressed within routines that are fun and involve family and/or friends.
Participation in daily life is further enhanced by using adaptive equipment to modify the environment.
At High Hopes, we follow a multidisciplinary approach to help enhance the quality of life for children with life-limiting conditions, using equipment to enhance abilities and facilitate participation in age-appropriate and meaningful activities and life experiences.